Molecular insights into cucurbit[8]uril-mediated complexes: Enhanced interaction cooperation towards pseudostatic dynamics
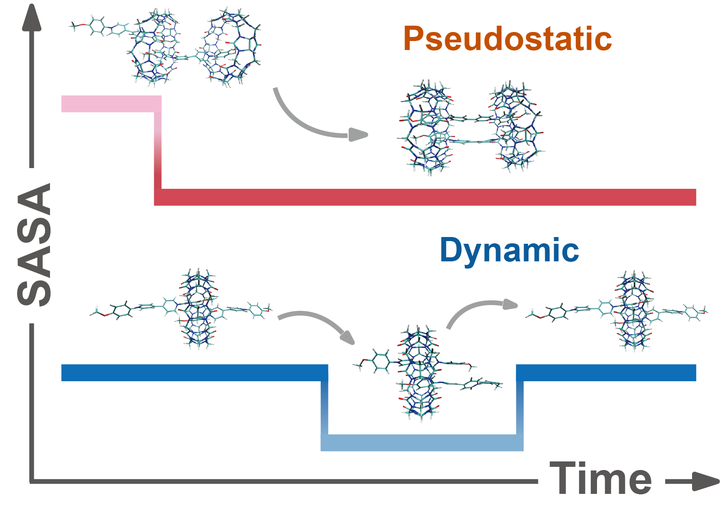 Image credit: ELSEVIER
Image credit: ELSEVIER摘要
Cucurbit[8]uril-mediated complexes are widely investigated due to their high binding affinity and diverse binding modes. However, the dynamic variations observed among different binding modes have yet to be fully understood, and current instrumental analyses impose temporal and spatial restrictions on the elucidation of instantaneous intra-complex motions. In this work, we performed molecular dynamics (MD) simulations and quantum chemical calculations on a CB[8]-diarylviologen complex system. Our simulations successfully monitored instantaneous intra-complex motions across multiple binding species using solvent accessible surface area (SASA) as the descriptor, which for the very first time provided a direct proof of the pseudostatic behavior. Additionally, we found that enhanced interaction cooperation resulted in the 2:2 complex being more energy-favorable than the 1:1 counterpart despite the same stoichiometry. These findings offer insights into the variable dynamics of host-guest complexes and the crucial role of interaction cooperation in controlling binding pathways, shedding a light on engineering supramolecular systems with greater precision.