 Image credit: Source
Image credit: Source摘要
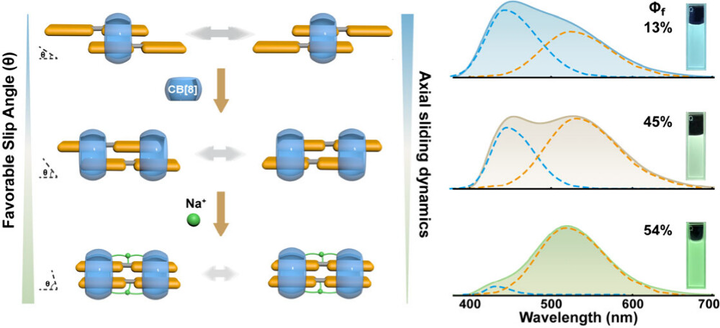
Control over luminescent properties is conventionally achieved by designing rigid, static packing geometries. Yet, chromophores within these assemblies naturally undergo continuous relative motion; harnessing this often-overlooked dynamic flexibility to actively dictate excited-state outcomes offers a powerful new dimension in materials design. Here, we introduce a supramolecular strategy to systematically control dual emission by restricting the structural dynamics of macrocycle-confined dimers. Utilizing cucurbit[8]uril (CB[8]) macrocyclic host and bis(phenylpyridinium) (BPP) guests, we construct precise 2:1 and 2:2 host–guest complexes to establish dynamic and static mobility limits within a unified framework. Cavity-confined dimerization induces a unique intrinsic dual emission. By progressively tightening structural restriction─moving from the fluxional 2:1 complex to the clamped 2:2 architecture, and further to a rigidly sodium-bridged framework─the dominant emission cleanly shifts from a short-wavelength state to a long-wavelength state, accompanied by a dramatically enhanced fluorescence quantum yield. Time-resolved spectroscopy reveals that this supramolecular confinement actively governs the kinetics of excited-state relaxation, definitively linking motional freedom to the resulting functional photoluminescence. Collectively, these results showcase the controlled restriction of supramolecular dynamics as an innovative, general design principle for tailoring programmable optoelectronic materials.